Aplicaciones exitosas
Aplicaciones exitosas
del diseño de fármacos utilizando
métodos computacionales 
José Luis Medina Franco
Los fármacos actúan produciendo cambios en algún proceso o función fisiológica. Muchos ejercen su efecto al interactuar específicamente con alguna estructura macromolecular del organismo. Para referirse a esta macromolécula, Paul Erlich propuso el término “receptor” a principios del siglo pasado. De esta forma, la interacción de cada fármaco con su respectivo receptor o sitio de acción inicia los cambios bioquímicos y fisiológicos que son característicos de ese fármaco.
Un ejemplo de un sitio de acción son los de las enzimas, proteínas especializadas en acelerar reacciones bioquímicas. La interacción de un fármaco con su blanco molecular se ha comparado en forma muy sencilla con lo que sucede entre una llave y una cerradura. En este caso, la llave encaja en la cerradura para producir una acción: abrir o cerrar esa cerradura.
Cuando se realiza la investigación de nuevos fármacos, nuevos o modificados a partir de moléculas existentes, primero se recopila toda la información posible sobre los procesos biológicos asociados a la enfermedad bajo estudio. En diversas ocasiones se logra identificar a un blanco molecular específico sobre el cual puede actuar un fármaco. Posteriormente se trata de determinar la estructura tridimensional de ese blanco para conocer con detalle el sitio de unión sobre el cual se pretende que interactúe el fármaco. Volviendo a la analogía de la llave con la cerradura, si se quiere fabricar una llave que entre en una cerradura determinada, esto será más sencillo si se conoce la forma de la cerradura.
 Una de las técnicas más empleadas actualmente para conocer la estructura tridimensional de macromoléculas es la cristalografía de rayos X. Otra técnica experimental es la resonancia magnética nuclear. A pesar de que el conocimiento de la estructura tridimensional es muy valioso para entender los mecanismos de acción de fármacos y diseñar a nuevas moléculas, en muchas ocasiones ésta se desconoce. Esto se debe, en parte, a la dificultad que presenta el uso de este tipo de técnicas. En estos casos, se puede recurrir a métodos computacionales, por ejemplo, a la metodología conocida como “modelado por homología”.
Una de las técnicas más empleadas actualmente para conocer la estructura tridimensional de macromoléculas es la cristalografía de rayos X. Otra técnica experimental es la resonancia magnética nuclear. A pesar de que el conocimiento de la estructura tridimensional es muy valioso para entender los mecanismos de acción de fármacos y diseñar a nuevas moléculas, en muchas ocasiones ésta se desconoce. Esto se debe, en parte, a la dificultad que presenta el uso de este tipo de técnicas. En estos casos, se puede recurrir a métodos computacionales, por ejemplo, a la metodología conocida como “modelado por homología”.
Los grupos de investigación alrededor del mundo que determinan la estructura tridimensional de macromoléculas almacenan sus resultados en la base de datos pública llamada Protein Data Bank (Banco de Datos de Proteínas, ver “páginas en internet de interés” al final del artículo). Actualmente, esta base de datos contiene información de aproximadamente cuarenta y un mil estructuras, y se actualiza constantemente. Dentro de esta base de datos cada estructura tridimensional tiene asociado un código de cuatro caracteres (llamado código PDB) que la identifica en forma única. Para la visualización en tercera dimensión de estas macromoléculas hay diversas herramientas en internet a las que se puede acceder desde la misma página del Protein Data Bank. También hay diversos programas computacionales gratuitos (ver “páginas en internet de interés”). Algunos de estos programas permiten incluso realizar cierto manejo de las estructuras y cálculos teóricos.
Al uso de la información estructural del blanco molecular para diseñar fármacos se le llama “diseño de fármacos basado en la estructura del receptor”. En este tipo de diseño las computadoras son una herramienta muy valiosa; casi imprescindible. Los programas computacionales permiten no solamente visualizar las estructuras de macromoléculas sino también hacer cálculos teóricos y predicciones de la afinidad que tendrá una molécula, aun hipotética, con el sitio de unión. Esta capacidad de predicción ayuda a proponer hipótesis y planear experimentos para hacerlos más enfocados y sistemáticos. La importancia y gran uso de las computadoras en el diseño de fármacos ha dado origen al área de investigación conocida como “diseño de fármacos asistido por computadora”.
Diseño de fármacos basado en la
estructura del receptor
Se define como la búsqueda de moléculas que encajen en el sitio de unión de un blanco molecular de manera que puedan formar interacciones favorables. Uno de los aspectos más importantes del diseño basado en la estructura del receptor busca mejorar las interacciones que ocurren entre una molécula y su blanco molecular. Esto es, se parte de un fármaco conocido pero que no tiene el efecto requerido; sin embargo, la estructura tridimensional del complejo molécula-blanco proporciona información para realizar la optimización. Con este propósito se recurre a uno de los métodos computacionales más usados en este campo: el “acoplamiento molecular automatizado” (Kitchen y colaboradores, 2004). Este método consiste en calcular con la computadora cuál es la posición más favorable que tendría una molécula con el blanco molecular. A partir del resultado se propocho, se puede trabajar con moléculas hipotéticas que no se tienen en el laboratorio, o que aún no han sido preparadas. En la industria farmacéutica es común trabajar con las llamadas “bibliotecas virtuales”: colecciones de miles o millones de moléculas hipotéticas. Sin embargo, después de hacer los cálculos, estas moléculas virtuales pueden ser fabricadas y evaluadas biológicamente.
 Otro aspecto del diseño basado en la estructura del receptor es diseñar, desde cero, una molécula que tendrá interacciones favorables con el blanco molecular. A este proceso se le conoce como “diseño de novo” y ha tenido avances notables en el diseño de fármacos (Schneider y Fechner, 2005). En general, esta estrategia consiste en construir, fragmento a fragmento, una molécula que según los cálculos tendrá interacciones favorables con el receptor. Los probables sitios de unión pueden conocerse a través de experimentos (por ejemplo, mutaciones dirigidas o resonancia magnética nuclear) o con la ayuda de métodos computacionales.
Otro aspecto del diseño basado en la estructura del receptor es diseñar, desde cero, una molécula que tendrá interacciones favorables con el blanco molecular. A este proceso se le conoce como “diseño de novo” y ha tenido avances notables en el diseño de fármacos (Schneider y Fechner, 2005). En general, esta estrategia consiste en construir, fragmento a fragmento, una molécula que según los cálculos tendrá interacciones favorables con el receptor. Los probables sitios de unión pueden conocerse a través de experimentos (por ejemplo, mutaciones dirigidas o resonancia magnética nuclear) o con la ayuda de métodos computacionales.
Aplicaciones exitosas del diseño
de fármacos asistido por computadora
Para muchas enfermedades se conocen estructuras tridimensionales de potenciales sitios de acción de fármacos. En diversas ocasiones los cálculos computacionales han tenido un papel muy importante en la investigación de moléculas que se unen a estos blancos y que actualmente se encuentran en uso clínico. Por ejemplo, el diseño de fármacos asistido por computadora ya ha tenido contribuciones notables en el tratamiento del síndrome de la inmunodeficiencia adquirida, en infecciones por el virus de la influenza y en el tratamiento del glaucoma.
Captopril: primer diseño basado en el receptor
La llamada enzima convertidora de angiotensina participa en dos procesos que parecen ser importantes en la regulación presión arterial. Por una parte, acelera la conversión de la angiotensina I en angiotensina II, que es un vasoconstrictor potente. Por otra parte inactiva a la bradiquinina, que es un vasodilatador. Por tanto, la enzima convertidora de angiotensina es un blanco molecular adecuado para el tratamiento de pacientes con hipertensión.
La investigación de inhibidores de la enzima convertidora de angiotensina surgió a partir de una sustancia natural obtenida de una víbora venenosa del Brasil. Al momento de iniciarse los estudios, en la década de los setenta, no se conocía la estructura tridimensional de esta enzima. Sin embargo, sí se conocía la estructura de una enzima parecida, la carboxipeptidasa A de bovino. Utilizando a esta estructura como modelo y siguiendo una metodología que actual-mente se denomina “diseño de análogo activo”, investigadores de la compañía Squibb desarrollaron el captopril, aprobado en Estados Unidos para su uso clínico en 1981. Aunque el captopril no se desarrolló utilizando cálculos computacionales se puede considerar como el primer ejemplo de un fármaco diseñado basado en la estructura del receptor. Recientemente se dio a conocer la estructura del captopril unido a la enzima convertidora de angiotensina (Figura 1), confirmando la hipótesis bajo la cual fue diseñado.
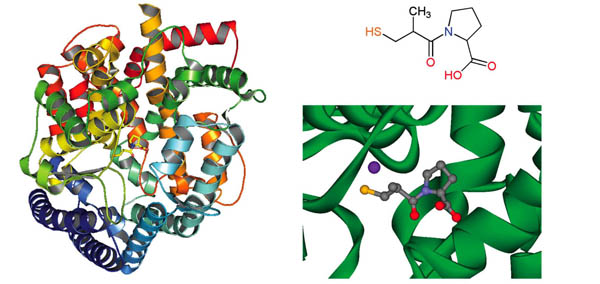
Figura 1. Estructura tridimensional de la enzima convertidora de angiotensina (izquierda) (código PDB: 1UZF), estructura química del captopril (derecha arriba) y detalle de la unión con la enzima.
Fármacos para el tratamiento del glaucoma
La anhidrasa carbónica II humana es una enzima que facilita la hidratación del dióxido de carbono para formar bicarbonato. Aunque diversas anhidrasas carbónicas se localizan en varios órganos, tejidos y células del cuerpo, la anhidrasa carbónica II es de especial interés porque su actividad está asociada a un incremento en la presión intraocular, produciendo de esta manera el glaucoma. Por lo tanto, los inhibidores de la anhidrasa carbónica son atractivos para el tratamiento de esta enfermedad.
Desde hace muchos años se ha utilizado a la metazolamida para el tratamiento del glaucoma. Sin embargo, debido a que este fármaco se administra en forma oral, causa efectos secundarios al inhibir la anhidrasa carbónica que se encuentra en otras partes del cuerpo. Por esta razón, era deseable contar un fármaco que pudiese administrarse en forma tópica (local y externa).
A mediados de la década de los ochenta la compañía Merck obtuvo por cristalografía de rayos X la estructura de la anhidrasa carbónica unida a una molécula que denominaron MK-927. A partir de esta estructura, de cálculos de la energía de diversas moléculas similares a la MK-927 y de estudios cristalográficos, se diseñó a la dorzolamida (Figura 2), un inhibidor potente de la anhidrasa carbónica. Su uso clínico se aprobó en 1994, con el nombre de Trusopt® y fue el primer inhibidor de la anhidrasa carbónica que se logró formular como solución oftálmica y, por tanto, administrarse en forma tópica. Cuatro años después se aprobó a la brinzolamida (Azopt®) que tiene una estructura química muy parecida a la dorzolamida y también se administra en forma tópica (Figura 2).
Fármacos para el tratamiento
del glaucoma
Otro ejemplo del éxito del diseño de fármacos utilizando la estructura del receptor y métodos computacionales es el desarrollo del relenza, fármaco empleado para el tratamiento de la infección causada por el virus de la influenza. Utilizando la estructura tridimensional de la enzima sialidasa (llamada también neuraminidasa, un blanco molecular para atacar al virus), y métodos computacionales, se propuso la estructura de inhibidores potentes de esta enzima (von Itzstein y colaboradores, 1993). La estrategia computacional consistió principalmente en el análisis gráfico de la estructura de la sialidasa y el uso del programa GRID. Este programa, muy utilizado en el diseño de novo de fármacos, es especialmente valioso para encontrar posibles sitios de unión en una macromolécula. Uno de los compuestos diseñados en la compañía hoy llamada GlaxoSmithKline, fue el relenza que se aprobó para su uso clínico en 1999 con el nombre de Zanamivir® (Figura 3). Otro fármaco para el tratamiento de la influenza es el oseltamivir (Tamiflu®), también aprobado en 1999 y hoy comercializado por la compañía Roche. La investigación de este fármaco, que está relacionado con el relenza, también se hizo apoyándose en la estructura tridimensional de la sialidasa.
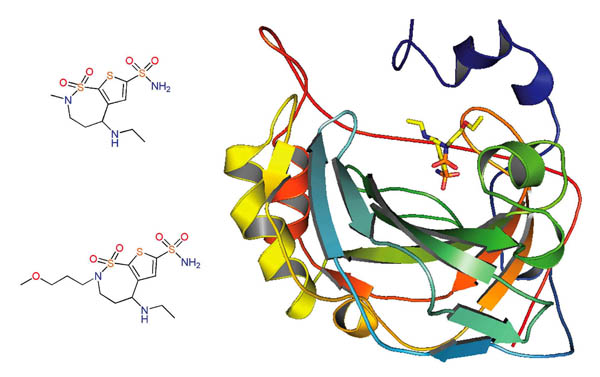
Figura 2. Estructura química de la dorzolamida (Trusopt ) (izquierda mensional de la anhidrasa carbónica (código PDB: 1A42).
Fármacos contra el SIDA
El mayor número de aplicaciones exitosas del diseño basado en la estructura del receptor con la ayuda de métodos computacionales ha ocurrido hasta ahora en el campo del tratamiento del síndrome de la inmunodeficiencia adquirida (sida). Poco después de que se detectaran los primeros casos a principios de la década de los ochenta, se encontró que el virus de la inmunodeficiencia humana (VIH) es el causante de esta enfermedad.
Hay diversos blancos moleculares sobre los cuales pueden interactuar fármacos para detener la infección causada por este virus. Uno de ellos es la enzima proteasa del VIH, que interviene en la maduración de las partículas virales. La estructura tridimensional de esta enzima se dio a conocer a finales de la década de los ochenta. Hacia 1990 se reportó una de las primeras aplicaciones del diseño basado en la estructura de esta enzima con el desarrollo del compuesto entonces llamado Ro 31-8959. Este diseño culminó cinco años después con la aprobación del saquinavir como el primer inhibidor de la proteasa del VIH utilizado en el tratamiento del sida (Figura 4). A partir de la estructura tridimensional de esta enzima se han diseñado y aprobado para su uso clínico ocho inhibidores de la proteasa de VIH. El inhibidor de más reciente aprobación es el tipranavir (Aptivus®), cuyo uso clínico se autorizó en Estados Unidos el 22 de junio de 2005 (Figuras 4 y 5). El uso de métodos computacionales, aunado al análisis estructural y síntesis química, ha participado en forma muy importante en la investigación que produjo estos fármacos. Los estudios computacionales han involucrado, generalmente, análisis gráficos de las estructuras tridimensionales y cálculos de energía. Resulta muy interesante el desarrollo del indinavir (Figura 4, referido originalmente por la compañía Merck con el código L-735,524), que involucró la predicción correcta de la actividad biológica de diversas moléculas utilizando cálculos teóricos (Holloway y colaboradores, 1995).
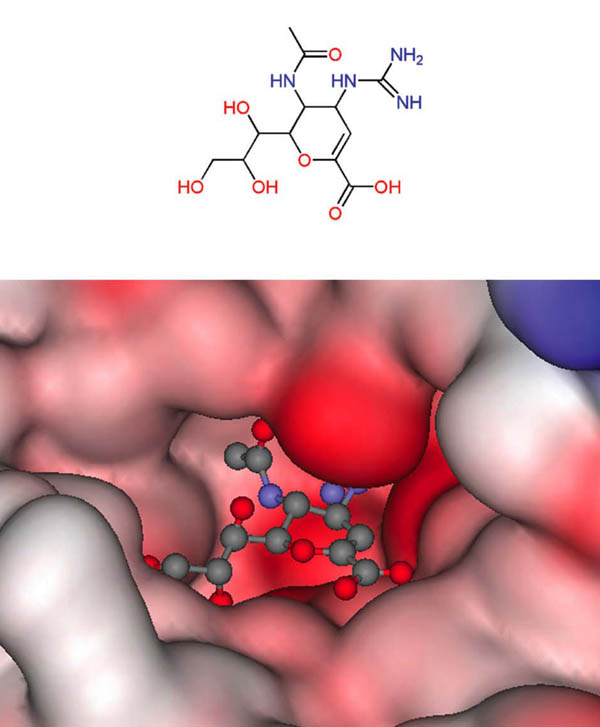
Figura 3. Estructura química del relenza (Zanamavir®) y detalle del sitio de unión con la enzima sialidasa del virus de la influenza (código PDB: 1A4G).
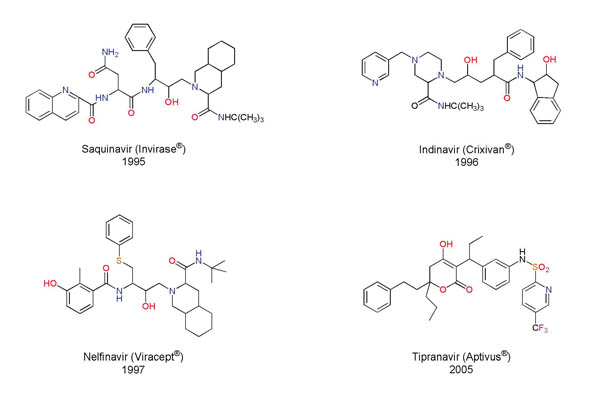
Figura 4. Ejemplos de fármacos empleados en el tratamiento del sida que inhiben a la proteasa del VIH. Se muestra el nombre comercial entre paréntesis y el año de aprobación.
Otro de los blancos moleculares de gran interés para atacar al virus del sida es la enzima transcriptasa inversa. Esta enzima, que convierte el ácido ribonucleico (ARN) que forma los genes del VIH en ácido desoxirribonucleico (ADN), participa en la replicación de las partículas virales dentro de la célula huésped. En México se está trabajando, mediante técnicas computacionales, en el diseño basado en la estructura del receptor de inhibidores de esta enzima. Utilizando acoplamiento molecular automatizado se han estudiado las interacciones que puede haber entre diversas moléculas y la transcriptasa inversa del VIH (Medina-Franco y colaboradores, 2004a). Partiendo de estos cálculos y otros estudios computacionales se han diseñado moléculas que son inhibidores prometedores de este blanco molecular (Medina-Franco y colaboradores, 2004b).
El futuro del diseño de fármacos mediante técnicas computacionales
Además de los fármacos diseñados con la ayuda de métodos computacionales se ha determinado la estructura tridimensional de diversos fármacos en uso clínico, obtenidos o diseñados por otros métodos, y de sus respectivos blancos moleculares. Algunos ejemplos son la aspirina unida a la enzima cycloooxigenasa I (aunque se sabe que también se une a la cycloooxigenasa II; de ahí sus efectos secundarios de irritación del estómago, gastritis y daño renal), el imatinib (Gleevec®) empleado en el tratamiento de ciertos tipos de enfermedades malignas (específicamente la leucemia mieloide crónica y los tumores del estroma gastrointestinal) y la atorvastatina (Lipitor®), usada en el tratamiento de la hiperlipidemia. Esta información está siendo de gran utilidad para el diseño de nuevos fármacos.
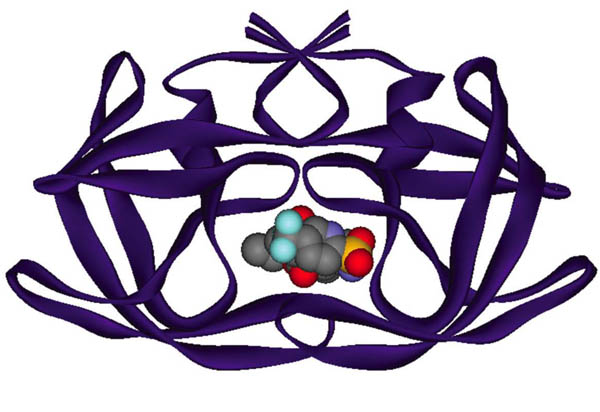
Figura 5. Estructura de la proteasa del VIH unida al tipranavir (Aptivus®) (código PDB: 1D4S).
Hoy el uso de la información estructural de los blancos moleculares y métodos computacionales es una práctica común en la industria farmacéutica y en instituciones académicas y de gobierno de todo el mundo. Esto ha sido favorecido por el fácil acceso a muchos programas que son potentes y tienen un costo muy bajo o incluso son gratuitos. Asimismo, la capacidad de los equipos de cómputo va en un aumento vertiginoso, y los costos de estos equipos son cada vez más accesibles. Como consecuencia, además de los ejemplos mostrados de diseño exitoso de fármacos con métodos computacionales, hay muchos casos en que estas metodologías están aportando información muy importante a la investigación. En México, además de los estudios mencionados con la transcriptasa inversa, se ha realizado acoplamiento molecular automatizado con la isomerasa de triosasfosfato (Espinoza-Fonseca y Trujillo-Ferrara, 2004) y con la 3-hidroxi-3-metilglutaril-coenzima A reductasa (Medina-Franco y colaboradores, 2005) para contribuir en el diseño de compuestos antiparasitarios para combatir el colesterol en la sangre (hipocolesterolemiantes), respectivamente. Otras aplicaciones se extienden a diversas enfermedades incluyendo el cáncer, el sida, la enfermedad de Alzheimer, la hipertensión y la artritis, entre muchas otras. Estas aplicaciones, así como los avances en el desarrollo de métodos y programas computacionales para el diseño de fármacos, pueden encontrarse en artículos publicados en revistas como Journal of Medicinal Chemistry, Journal of Computer-Aided Molecular Design, Journal of Chemical Information and Modeling, Bioorganic and Medicinal Chemistry, Current Computer-Aided Drug Design, Nature Reviews Drug Discovery y Science, entre otras. Sin duda, en un futuro cercano veremos más fármacos desarrollados con la ayuda de métodos computacionales.
Conclusión
Las computadoras se han convertido en una poderosa herramienta para generar modelos de fármacos interactuando con biomoléculas. Estos modelos ayudan a entender el mecanismo de acción de los fármacos, proponer modificaciones para mejorar su efecto terapéutico y diseñar nuevas moléculas. Hoy en día, ya hay diversos fármacos en uso clínico que fueron diseñados con la ayuda de métodos computacionales, muchos de ellos para el tratamiento del sida.
Agradecimientos
Agradezco a la doctora Karina Martínez (Universidad de Arizona) y al doctor Heriberto Medina (Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán) sus valiosos comentarios. Dedico este trabajo a la Fundación Lorena Alejandra Gallardo en sus veinticinco años de trabajo incansable en el apoyo a estudiantes mexicanos.
 Para saber más
Para saber más
Espinoza-Fonseca, L. M. y J. G. Trujillo-Ferrara (2004), “Exploring the possible binding sites at the interface of triosephosphate isomerase dimer as a potential target for anti-tripanosomal drug design”, Bioorganic and Medicinal Chemistry Letters, 14, 3151-3154.
Gohlke, H. y G. Klebe (2002), “Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors”, Angewandte Chemie International Edition, 41, 2644-2676.
Holloway, M. K. y colaboradores (1995), “A priori prediction of activity for HIV-1 protease inhibitors employing energy minimization in the active site”, Journal of Medicinal Chemistry, 38, 305-317.
Jorgensen, W. L. (2004), “The many roles of computation in drug discovery”, Science, 303, 1813-1818.
Kuntz, I. D. (1992), “Structure-based strategies for drug design and discovery”, Science, 257, 1078-1082.
Kitchen, D. B. y colaboradores (2004), “Docking and scoring in virtual screening for drug discovery: methods and applications”, Nature Reviews Drug Discovery, 3, 935-949.
Medina-Franco, J. L. y colaboradores (2004a), “Docking-based CoMFA and CoMSIA studies of non-nucleoside reverse transcriptase inhibitors of the pyridinone derivative type”, Journal of Computer-Aided Molecular Design, 18, 345-360.
Medina-Franco, J. L. y colaboradores (2004b), “Flexible docking of pyridinone derivatives into the non-nucleoside inhibitor binding site of HIV-1 reverse transcriptase”, Bioorganic and Medicinal Chemistry, 12, 6085-6095.
Medina-Franco, J. L. y colaboradores (2005), “Molecular docking of the highly hypolipidemic agent ?-asarone with the catalytic portion of HMG-CoA reductase”, Bioorganic and Medicinal Chemistry Letters, 15, 989-994.
Schneider, G. y U. Fechner (2005), “Computer-based de novo design of drug-like molecules”, Nature Reviews Drug Discovery, 4, 649-663.
von Itzstein, M. y colaboradores (1993), “Rational design of potent sialidase-based inhibitors of influenza virus replication”, Nature, 363, 418-423.
Páginas en internet de interés
Protein Data Bank, banco de datos de blancos moleculares y otras estructuras tridimensionales: http://pdbbeta.rcsb.org
Algunos programas gratuitos para visualizar estructuras moleculares en tercera dimensión:Chimera:
http://www.cgl.ucsf.edu/chimera/;
Molscript:
http://www.avatar.se/molscript/;
Pymol:
http://pymol.sourceforge.net/;
Raster 3D:
http://www.bmsc.washington.edu/raster3d/. (En la página del Protein Data Bank puede accederse directamente a los visualizadores King Viewer, Jmol Viewer, WebMol Viewer, entre otros.)
Programa de cómputo gratuito para dibujar estructuras químicas: ACD Labs Chemsketch: http://www.acdlabs.com/download/
Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo.